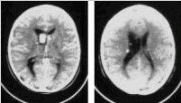
Tuberous sclerosis A 6 year-old male patients, child of non-consanguineous parents with no family history of neurometabolic or neurodegenerative disorders opresented to the Emergency Department because of sudden-onset neurologic symptomatology with irritability, vomiting, gaze paresis, gate disturbance, seizures and drowsiness rapidly progressing to stupor. At presentation he had a body temperature of 37.5 °C, a blood pressure of 105/75 mmHg, a respiratory rate of 40 breaths/min and a pulse rate of 65 beats/min. There was no heart murmur and the auscultation of the chest demonstrated clear breath sounds bilateraly and normal heart sounds without a murmur. Length, weight and head circumference were within normal limits. No organomegaly was present. Similarly, no signs of infection were present. The rest of the physical examination was unremarkable except for three oval shaped, lightly pigmented, presumably hypomelanotic macules, visible with naked eye, one over patient’s right thigh (fig. 1) and the others at the back and at left buttock, respectively. No café au-lait macules or other pigment anomalies were noted. Neurological examinaion demonstrated increased deep tendon reflexes (left>right), as well as left lower extremity clonus, Babinski’s sign and Rossolimo’s relfex, wel preserved pupillary reaction to light, left abducents palsy and a score of 9 in the Glasgow Coma Scale. CT of the brain fig. 2) was diagnostic of the patient’s condition.
|
 TS is an autosomal dominant neurocutaneous syndrome
characterized by a wide variety of neurologic symptoms and small tumors
of the brain, kidneys, liver, spleen and lungs. Brain findings include
cortical and subcortical tubers, subependymal nodules and SEGAs typically
located around the foramen of Monro; typical clues to diagnosis are the
characteristic skin manifestations like hypomelanotic macules, shagreen
patches, facial angiofibromas and periungual fibromas. Affected individuals
may be of normal intelligence or developmen-tally retarded, usually suffering
from an intractable epilepsy presenting during the first year of life
as in-fantile spasms. The characteristic skin finding of adenoma sebaceum
does not appear until 2 to 6 years of age and is not unusual for the hypopigmented
macules not to be the cause of parental concern, unless developmental
retardation or bad school performance lead to a pediatric consultation.
Although TS mani-fests itself most often subacutely, sudden-onset symptomatology
with focal neurologic signs, seizures and signs of increased intracranial
pressure can be the initial presentation of a hitherto undiagnosed older
TS patient, due to SEGA resulting in occlusion of the foramen of Monro
and to subsequent ventricular dilata-tion. It must be kept in mind that
intermittent symptoms, like headaches, vomiting or extreme sleepiness,
can be present in such a patient long before the SEGAs result to permanent
occlusion of the foramen of Monro, and are misdiagnosed as tension headaches
or migraines.
TS is an autosomal dominant neurocutaneous syndrome
characterized by a wide variety of neurologic symptoms and small tumors
of the brain, kidneys, liver, spleen and lungs. Brain findings include
cortical and subcortical tubers, subependymal nodules and SEGAs typically
located around the foramen of Monro; typical clues to diagnosis are the
characteristic skin manifestations like hypomelanotic macules, shagreen
patches, facial angiofibromas and periungual fibromas. Affected individuals
may be of normal intelligence or developmen-tally retarded, usually suffering
from an intractable epilepsy presenting during the first year of life
as in-fantile spasms. The characteristic skin finding of adenoma sebaceum
does not appear until 2 to 6 years of age and is not unusual for the hypopigmented
macules not to be the cause of parental concern, unless developmental
retardation or bad school performance lead to a pediatric consultation.
Although TS mani-fests itself most often subacutely, sudden-onset symptomatology
with focal neurologic signs, seizures and signs of increased intracranial
pressure can be the initial presentation of a hitherto undiagnosed older
TS patient, due to SEGA resulting in occlusion of the foramen of Monro
and to subsequent ventricular dilata-tion. It must be kept in mind that
intermittent symptoms, like headaches, vomiting or extreme sleepiness,
can be present in such a patient long before the SEGAs result to permanent
occlusion of the foramen of Monro, and are misdiagnosed as tension headaches
or migraines.